Neurodegeneration
Mitochondrial dysfunction in neurodegeneration
Mitochondrial dysfunction is an early event in the pathogenesis of neurodegenerative diseases. Reduced ATP levels, increased reactive oxygen species (ROS), impaired calcium buffering, and altered mitochondrial permeability are characteristic mitochondrial defects (Lin & Beal, 2006) . In many neurodegenerative disorders, specific subsets of neurons degenerate while others are unaffected (Bossy-Wetzel et al., 2008). Despite the clear association of mitochondrial dysfunction with neurodegenerative disease the molecular origins of these neuroselective pathologies are poorly understood.
A C. elegans model for neurodegeneration
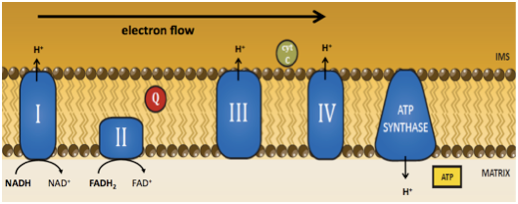
Coenzyme Q provides essential functions in the mitochondrial electron transport chain (ETC). We have shown that depletion of Coenzyme Q (CoQ) through RNAi and genetic ablation of CoQ biosynthetic genes, coq-1, coq- 2, and coq-3, leads to the progressive loss of motor coordination and preferential degeneration of GABA neurons (Earls & Hacker et al. 2010). The mechanism of cell death relies strongly on the function of ced-4 (Apaf-1) and partially depends on ced-3 (caspase) activity. The GABA neuron pathology that accompanies CoQ depletion also involves calcium signaling, possibly from ER stores, and the mitochondrial fission protein DRP-1. These results emphasize an important role for CoQ in neuron survival and link mitochondrial dysfunction to a calcium-dependent mechanism of selective neuron degeneration in C. elegans.