Research
Quantitative systems biology of dynamic signal transduction and gene regulation in single cells.
Lab summary
Long noncoding RNA biology
Signaling in physiological environments in human cells
A rate threshold mechanism regulates MAPK stress signaling and survival
Kinetics of osmotic stress regulates a cell fate switch of cell survival
Predictive modeling and model-identification of signal transduction and gene regulation
Combining quantitative experiments with predictive modeling to understand cell signaling
Overview:
Numerous human maladies arise from alterations in cellular and molecular pathways stemming from exposure to pernicious environmental conditions. Alas, the lion’s share of biological investigations into the molecular changes that occur in diseased cells are carried out in environments that do not adequately replicate physiological conditions. Additionally, even individuals who are diagnosed with the same ailment may exhibit vastly dissimilar reactions to treatment, yet most studies assume that all cells of a given type behave uniformly. To surmount these limitations, the Neuert laboratory is dedicated to comprehending the fundamental mechanisms of signal transduction and gene expression in normal and diseased physiology within the context of kinetics and variability. A quantitative framework is employed to tackle broad biological inquiries, utilizing a diverse set of methods such as single-cell, single-molecule, and genome-wide assays, along with computational data analysis, genetics, molecular biology, chemical profiling, and single-cell predictive computational modeling. This integrative experimental and computational framework constitutes the bedrock of the Neuert lab’s expertise.
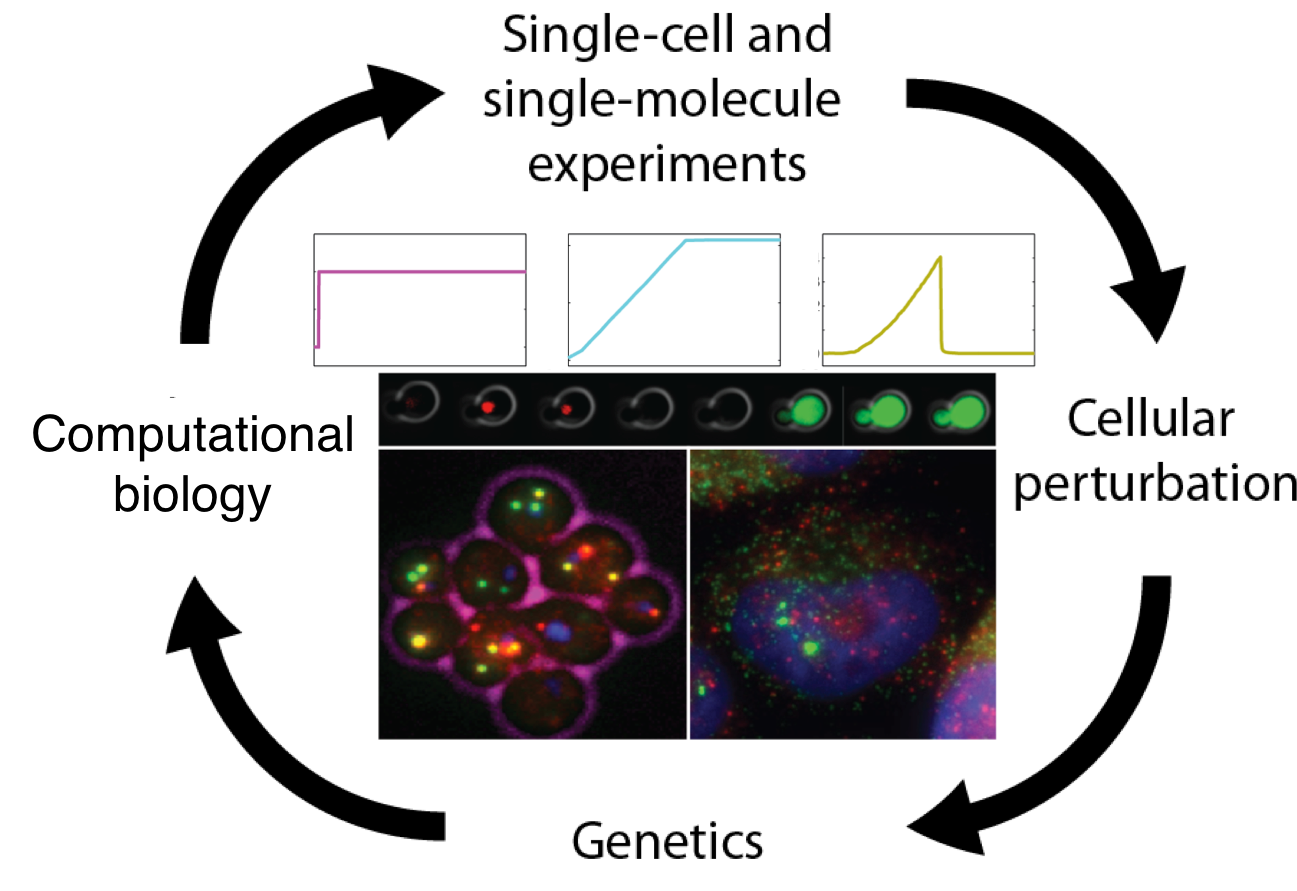
Research areas are:
Study signal transduction and gene regulation in physiologically relevant environments:
In a bid to explore signal transduction and gene regulatory pathways in physiologically appropriate environments, we have crafted various techniques that facilitate exacting environmental manipulation. Such methods have been successfully implemented to scrutinize and govern cellular stress response, leading to the capability to manipulate cellular phenotype, signal transduction, and gene regulation. Notably, these methodologies are universally applicable across biological pathways and organisms and are equally effective in studying a broad spectrum of signal transduction and gene expression pathways.Given that a multitude of intricate diseases are attributed to protein malfunction within signaling and gene networks, it stands to reason that deeper insights into these networks may bolster the development of more precise therapies. Thus, enhancing the ability to define these networks holds the potential to provide an improved comprehension of cellular pathways, which can serve as the bedrock for more efficacious targeted therapies.
Selected publications:
Understand the function of the non-coding genome:
Despite constituting a significant portion of the transcribed genome, the precise function of long non-coding RNAs (lncRNAs) remains elusive. Yet, the underlying goal of this emerging field is to unravel how the expression of lncRNAs influences cellular function, particularly in relation to gene regulation. To gain a better understanding of the biological relevance of lncRNAs, we are meticulously investigating their expression dynamics and regulatory effects on neighboring mRNAs across a spectrum of cell types. By assiduously controlling environmental conditions and measuring downstream effects on lncRNA and mRNA expression patterns, we aspire to unearth the mechanisms by which lncRNAs modulate gene expression.Our approach is universally applicable and can be employed to scrutinize any inducible lncRNA or gene, irrespective of the organism or cell type. Building on this, we plan to apply the knowledge garnered from studying these systems to explore similar systems that have been linked to human disease. Ultimately, by shedding light on the mechanisms by which lncRNAs regulate gene expression, we aim to unlock new avenues for the development of targeted therapies for various diseases.
Selected publications:
Revolutionize predictive model identification to gain biological insight:
Phenotypic variation is ubiquitous in biology, resulting from a plethora of genetic and environmental factors. Remarkably, even genetically identical cells, when placed in identical environments, exhibit variable phenotypes due to stochastic gene expression. To comprehend the underlying regulatory networks and the intricate relationships between genes, we are developing optimal experimental design methods that quantify the unique stochastic variability patterns of different biological systems. We are pushing the envelope on advanced image processing and experimental approaches that measure spatial-temporal expression patterns of multiple RNA species within individual cells. Using this data, we generate multidimensional probability distributions that aid in revealing the structure of gene regulatory networks.These cell-to-cell variability patterns serve as unique ‘fingerprints,’ offering key insights into the relationships between genes within regulatory networks. An added advantage of our approach is that it does not require genetic manipulation of the biological system being studied, making it especially useful for studying mammalian gene regulatory networks. By leveraging the knowledge garnered from our cutting-edge predictive model identification techniques, we aspire to revolutionize the field of biology and gain a deeper understanding of the complex interplay between genes and their regulatory networks.
Selected publications:
Develop robust and data-driven analysis pipelines for dynamic single-cell and genomic data sets:
In order to extract maximum biological insight from dynamic single-cell and genomic data sets, it is essential to have robust and data-driven analysis pipelines. Our research focuses on developing these pipelines with the aim of creating scalable and reproducible results, while making a minimum number of assumptions. We believe that the ability to generate reproducible data between biological replicas is equally important as the reproducibility of data analysis pipelines. Our pipelines are designed to provide robust results even when analyzed differently by different individuals at different labs. Our innovative approach includes the development of single-cell signal transduction analysis of time-lapse microscopy movies, counting of long noncoding RNA in single cells, and quantification of nascent transcription in single cells. These pipelines are examples of our efforts to advance the field of dynamic single-cell and genomic data analysis.
Selected publications: