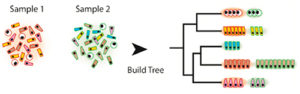
 Our growing ability to acquire data at the level of single cells in complex samples provides exciting new opportunities to understand physiological and pathophysiological processes; however, it also presents daunting challenges for the statistical evaluation of the resultant huge datasets. To date, most experimental approaches have focused on identifying the cell populations that are present in a single sample, but it is evident that much more can be gained by being able to evaluate changes in cell populations that occur in response to different conditions, such as exposure to a drug, presence of a pathogen, or normal physiological development. Current computational methods do not adequately enable such comparisons, leading Vanderbilt Basic Sciences investigators Ken Lau, his collaborator Qi Liu (Department of Biomedical Informatics) and their colleagues to develop sc-UniFrac to fill the void. Based on UniFrac, a previously published method designed to compare microbial cell populations, sc-UniFrac is designed to use single-cell RNAseq data to generate a hierarchical tree based on similarities and differences among the transcriptomes of the various cell populations in two or more samples of interest. A comparison of the trees between the samples enables calculation of the sc-UniFrac distance, a value based on the difference between the lengths of the branches in the trees weighted by the abundance of cells assigned to each branch. As the difference between the cell populations in the samples increases, so does the sc-UniFrac distance. The researchers showed that sc-UniFrac could distinguish between technical replicates (multiple evaluations of the same cell sample), biological replicates (evaluation of multiple samples of the same tissue origin), or distinct cell populations, with the sc-UniFrac distance increasing in that order, as would be expected. In addition, the method enabled the investigators to identify specific cell types that are responsible for the differences between populations. For example, in a comparison of colonic tumor tissue with surrounding normal epithelium, sc-UniFrac identified a population of granulocytes found only in the tumor as a result of inflammation. sc-UniFrac can be used to evaluate data of any statistical distribution, can be applied to other kinds of data, and can be used even if sample sizes are not the same. Thus, it appears that sc-UniFrac will be a valuable tool for the analysis of the increasingly complex datasets that will emanate from single cell experiments in the future. Thework is published in the journal PLoS Biology [Q. Liu, et al., (2018) PLoS Biol., 16, e2006687].
Our growing ability to acquire data at the level of single cells in complex samples provides exciting new opportunities to understand physiological and pathophysiological processes; however, it also presents daunting challenges for the statistical evaluation of the resultant huge datasets. To date, most experimental approaches have focused on identifying the cell populations that are present in a single sample, but it is evident that much more can be gained by being able to evaluate changes in cell populations that occur in response to different conditions, such as exposure to a drug, presence of a pathogen, or normal physiological development. Current computational methods do not adequately enable such comparisons, leading Vanderbilt Basic Sciences investigators Ken Lau, his collaborator Qi Liu (Department of Biomedical Informatics) and their colleagues to develop sc-UniFrac to fill the void. Based on UniFrac, a previously published method designed to compare microbial cell populations, sc-UniFrac is designed to use single-cell RNAseq data to generate a hierarchical tree based on similarities and differences among the transcriptomes of the various cell populations in two or more samples of interest. A comparison of the trees between the samples enables calculation of the sc-UniFrac distance, a value based on the difference between the lengths of the branches in the trees weighted by the abundance of cells assigned to each branch. As the difference between the cell populations in the samples increases, so does the sc-UniFrac distance. The researchers showed that sc-UniFrac could distinguish between technical replicates (multiple evaluations of the same cell sample), biological replicates (evaluation of multiple samples of the same tissue origin), or distinct cell populations, with the sc-UniFrac distance increasing in that order, as would be expected. In addition, the method enabled the investigators to identify specific cell types that are responsible for the differences between populations. For example, in a comparison of colonic tumor tissue with surrounding normal epithelium, sc-UniFrac identified a population of granulocytes found only in the tumor as a result of inflammation. sc-UniFrac can be used to evaluate data of any statistical distribution, can be applied to other kinds of data, and can be used even if sample sizes are not the same. Thus, it appears that sc-UniFrac will be a valuable tool for the analysis of the increasingly complex datasets that will emanate from single cell experiments in the future. Thework is published in the journal PLoS Biology [Q. Liu, et al., (2018) PLoS Biol., 16, e2006687].