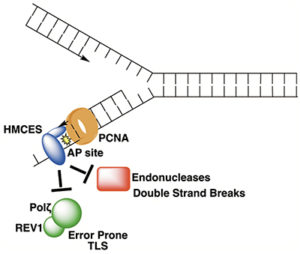
 In DNA, abasic sites (apurinic/apyrimidinic or AP sites) occur through spontaneous hydrolysis of the glycosidic bond that joins the base to the deoxyribose ring (a common reaction following formation of some kinds of DNA adducts) or through the action of DNA glycosylases during repair of damaged bases. In double-stranded DNA, AP sites are easily repaired using the complementary intact base as a template. However, such damage can also occur in single-stranded DNA (ssDNA), in which case no template is available to guide repair. Under these circumstances, attempted repair by translesion polymerases or AP endonucleases can lead to mutations or double-strand breaks, respectively. Now, Vanderbilt University Basic Sciences investigator David Cortez and his laboratory report the discovery that the protein HMCES (5-hydroxymethylcytosine-binding, embryonic stem cell-specific) binds to and protects ssDNA AP sites in replicating DNA. HMCES first came to the investigators’ attention through experiments using the iPOND (isolation of proteins on nascent DNA) technique to identify proteins present at sites of DNA replication. HMCES was one of the proteins they identified. Follow-up studies demonstrated that HMCES was most highly expressed during the S phase of the cell cycle, and that it translocated to the nucleus following exposure to DNA damaging agents. Cells bearing deletion mutations in the gene encoding HMCES were highly sensitive to inhibitors of the ATR checkpoint kinase. Together, these observations suggested that HMCES plays a role in DNA replication and/or repair, a hypothesis that was further supported by the finding that HMCES binds strongly to ssDNA and that it interacts strongly with PCNA (proliferating cell nuclear antigen), a protein that plays an important role in DNA replication. Cells bearing a deletion mutation in the gene encoding HMCES were hypersensitive to a group of DNA damaging agents that all lead to the formation of AP sites, and the researchers demonstrated that HMCES binds ssDNA containing an AP site with very high affinity. Further studies demonstrated that the interaction between HMCES and the AP site resulted from a covalent bond formed between the damaged DNA and a conserved cysteine residue in the protein. In fact, treating cells with an AP site-generating DNA damaging agent led to the formation of cross-links between DNA and HMCES. These cross-links accumulated in cells treated with a proteasome inhibitor, a finding that suggested that cross-linked HMCES is normally degraded by the proteasome. Consistently, exposure of cells to AP-generating DNA damaging agents led to ubiquitination of HMCES, thereby marking it for proteasomal degradation. Additional experiments revealed that binding of HMCES to AP sites in ssDNA configured as it would be found at a replication fork protected the site from cleavage by an AP endonuclease. The researchers also found that translesion polymerases accumulate at replication forks in HMCES-deficient cells, suggesting that these enzymes are recruited to compensate for the absence of HMCES. Accordingly, exposure of HMCES-deficient cells to AP site-generating DNA damaging agents led to higher levels of mutations than were seen in wild-type cells. Together, the results suggest that the main function of HMCES is to bind to and protect AP sites in ssDNA until repair can be accomplished. It is not completely clear how HMCES helps to mediate repair, but it is clear that its absence is deleterious to the cell’s ability to withstand exposure to DNA damaging agents. We await further work to gain a complete picture of how cells cope with AP sites in ssDNA now that we understand this initial protective response. The work is published in the journalCell [K. N. Mohni, et al. (2018) Cell, published December 13, DOI: 10.1016/j.cell.2018.10.055].
In DNA, abasic sites (apurinic/apyrimidinic or AP sites) occur through spontaneous hydrolysis of the glycosidic bond that joins the base to the deoxyribose ring (a common reaction following formation of some kinds of DNA adducts) or through the action of DNA glycosylases during repair of damaged bases. In double-stranded DNA, AP sites are easily repaired using the complementary intact base as a template. However, such damage can also occur in single-stranded DNA (ssDNA), in which case no template is available to guide repair. Under these circumstances, attempted repair by translesion polymerases or AP endonucleases can lead to mutations or double-strand breaks, respectively. Now, Vanderbilt University Basic Sciences investigator David Cortez and his laboratory report the discovery that the protein HMCES (5-hydroxymethylcytosine-binding, embryonic stem cell-specific) binds to and protects ssDNA AP sites in replicating DNA. HMCES first came to the investigators’ attention through experiments using the iPOND (isolation of proteins on nascent DNA) technique to identify proteins present at sites of DNA replication. HMCES was one of the proteins they identified. Follow-up studies demonstrated that HMCES was most highly expressed during the S phase of the cell cycle, and that it translocated to the nucleus following exposure to DNA damaging agents. Cells bearing deletion mutations in the gene encoding HMCES were highly sensitive to inhibitors of the ATR checkpoint kinase. Together, these observations suggested that HMCES plays a role in DNA replication and/or repair, a hypothesis that was further supported by the finding that HMCES binds strongly to ssDNA and that it interacts strongly with PCNA (proliferating cell nuclear antigen), a protein that plays an important role in DNA replication. Cells bearing a deletion mutation in the gene encoding HMCES were hypersensitive to a group of DNA damaging agents that all lead to the formation of AP sites, and the researchers demonstrated that HMCES binds ssDNA containing an AP site with very high affinity. Further studies demonstrated that the interaction between HMCES and the AP site resulted from a covalent bond formed between the damaged DNA and a conserved cysteine residue in the protein. In fact, treating cells with an AP site-generating DNA damaging agent led to the formation of cross-links between DNA and HMCES. These cross-links accumulated in cells treated with a proteasome inhibitor, a finding that suggested that cross-linked HMCES is normally degraded by the proteasome. Consistently, exposure of cells to AP-generating DNA damaging agents led to ubiquitination of HMCES, thereby marking it for proteasomal degradation. Additional experiments revealed that binding of HMCES to AP sites in ssDNA configured as it would be found at a replication fork protected the site from cleavage by an AP endonuclease. The researchers also found that translesion polymerases accumulate at replication forks in HMCES-deficient cells, suggesting that these enzymes are recruited to compensate for the absence of HMCES. Accordingly, exposure of HMCES-deficient cells to AP site-generating DNA damaging agents led to higher levels of mutations than were seen in wild-type cells. Together, the results suggest that the main function of HMCES is to bind to and protect AP sites in ssDNA until repair can be accomplished. It is not completely clear how HMCES helps to mediate repair, but it is clear that its absence is deleterious to the cell’s ability to withstand exposure to DNA damaging agents. We await further work to gain a complete picture of how cells cope with AP sites in ssDNA now that we understand this initial protective response. The work is published in the journalCell [K. N. Mohni, et al. (2018) Cell, published December 13, DOI: 10.1016/j.cell.2018.10.055].