
SWI/SNF is a multicomponent protein complex that plays an important role in chromatin remodeling. It is also likely an important tumor suppressor, as indicated by the fact that approximately 20% of human cancers carry a mutation in one or more SWI/SNF protein components. However, the exact mechanism by which these mutations contribute to cancer pathogenesis is not always clear, as is illustrated in the case of SNF5. This SWI/SNF component protein is mutated in a number of cancers, including malignant rhabdoid tumor (MRT), a highly aggressive, nearly uniformly fatal childhood cancer.
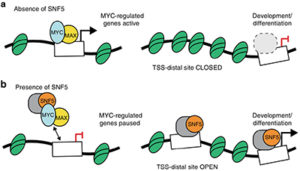
The fact that SNF5 is usually the only mutation found in MRT suggests that it plays an important role in tumorigenesis. However, in addition to serving as an SWI/SNF component, SNF5 can associate with the MYC oncoprotein and inhibit the binding of the MYC:MAX heterodimeric transcription factor to DNA. Thus, it is unclear whether the loss of SNF5 in MRT promotes malignancy through altering the function of MYC, SWI/SNF, or both. To answer this question, Vanderbilt Basic Sciences investigator William Tansey and his laboratory took a closer look at the interaction of SNF5 and MYC in MRT. They first used an electrophoretic mobility shift assay (EMSA) to show that SNF5 blocks binding of MYC:MAX to its E-box target sequence in DNA in vitro.
They then engineered HEK293 cells to enable them to control the level of SNF5 expression and demonstrated that an increase in SNF5 led to a decrease in MYC bound to nuclear DNA and vice versa. Expression of SNF5 in an MRT cell line reduced anchorage-independent growth of the cells and was similarly effective as expression of the OmoMYC dominant negative mutant at blocking the association of MYC with its target genes.
Using an assay that allowed them to assess the accessibility of DNA (a measure of chromatin structure), the researchers showed that SNF5 expression also increased accessibility of ~2,500 sites, whereas OmoMYC had no effect, consistent with SWI/SNF-mediated chromatin remodeling. These new sites, however, were distinct from those targeted by MYC, leading the researchers to conclude that the effects of SNF5 on MYC binding to DNA were not the result of SWI/SNF-mediated restructuring of chromatin. Finally, because MYC:MAX binding to target genes leads to a release of RNA polymerase II from a pause site at the proximal promoter of that gene, the researchers evaluated the effect of SNF5 on the location of RNA polymerase II in the genome.
They found that both SNF5 and OmoMYC increased the pausing of the polymerase at MYC-targeted genes in a similar fashion. Together the results demonstrate that an important means by which SNF5 suppresses tumorigenesis in MRT is through the blockade of MYC. This suggests that cancers bearing an SNF5 mutation may be susceptible to therapy by MYC inhibition, providing yet another reason why the discovery of clinically viable MYC inhibitors is of utmost importance. The work is published in the journal Nature Communications [A. M. Weissmiller, et al. (2019) Nat. Commun., 10, 2014].