
A team of Vanderbilt researchers has created a new series of drug candidates against a hard-to-target receptor involved in the formation of blood clots. The research, spearheaded by the labs of Jens Meiler, research professor of chemistry, Craig Lindsley, Executive Director of Warren Center for Neuroscience Drug Discovery and professor of pharmacology, and Heidi Hamm, the Aileen M. Lange and Annie Mary Lyle Professor of Cardiovascular Research and professor of pharmacology, was published in ACS Pharmacology and Translational Science in March 2024.



Blood clots that impede blood flow—thrombosis—can afflict anyone and can form in veins, such as in pulmonary embolisms, or arteries, such as in heart attacks or strokes. Although thrombosis is not necessarily lethal, the U.S. alone has seen 100,000 to 300,000 deaths per year related to blood clots prior to the onset of the pandemic, but that number is likely to have increased since. According to a 2022 population-wide study from the U.K., COVID-19 infection increases a person’s risk of blood clot-related problems immediately after infection and for up to 49 weeks afterward.
The risk of blood clots is aggravated by certain risk factors but can be mitigated through drugs such as antiplatelet drugs, but these cause undesirable side effects. Newer approaches to targeting platelets, which are key to the formation of blood clots, involve protease-activated receptor 4; recent data from Vanderbilt labs and others suggest that PAR4 may also be a viable drug target for inflammatory diseases such as kidney injury and Alzheimer’s disease.
However, despite its clear attraction, PAR4 is difficult to target. A cell surface receptor, PAR4 is activated when a protease called thrombin cleaves its tail, which exposes a “new” tail (called a tethered ligand) that interacts with other parts of PAR4. Since the receptor’s ligand is attached to the receptor itself, it has proven difficult to create a PAR4 inhibitor that can outcompete the tethered ligand and inhibit PAR4.
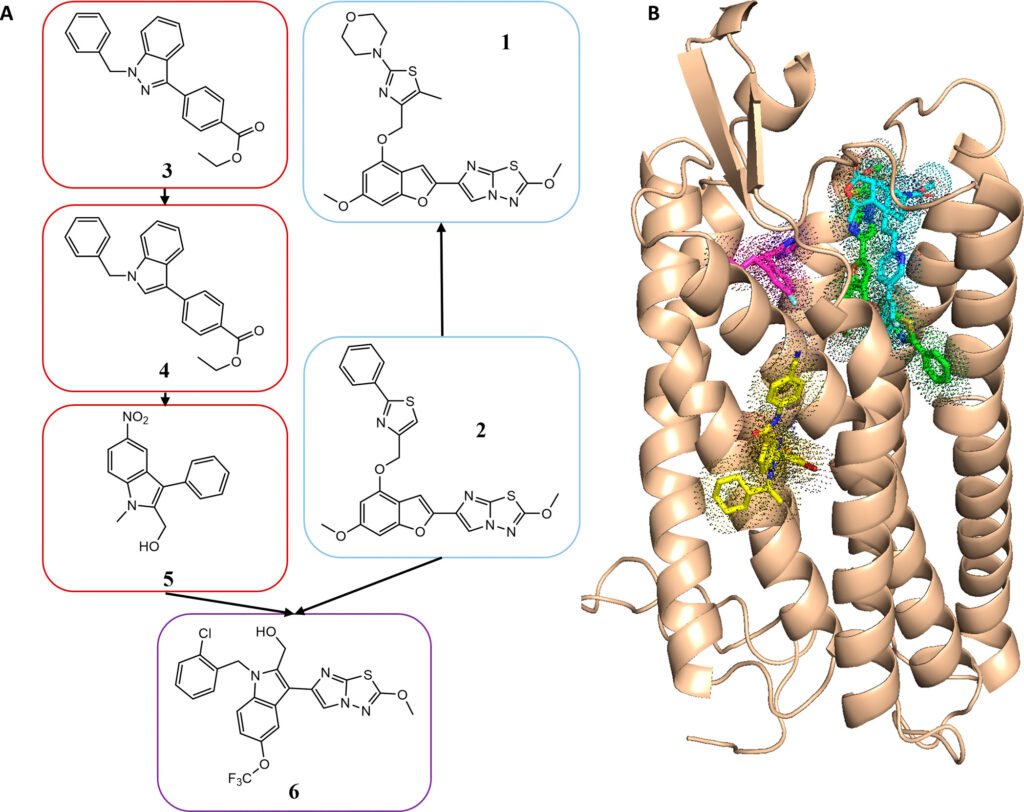
“The main technical innovation came from the application of ultra-large library screening to computer-aided drug design, an approach that allowed us to test billons of drug-like molecules in silico for biological activity,” said Shannon Smith, former graduate student in the Meiler laboratory and first author on the paper.
The authors used computer simulations to “dock” 129 million compounds to a computer model of PAR4. The novel antagonists identified in the screen were then improved through medicinal chemistry in the Lindsley laboratory and were tested for biological inhibition of the receptor. Although the use of this type of screen is not novel, such a screen had never been carried out with a protein without a known structure—only with proteins with known crystal structures.
“As no experimental structure of PAR4 was available at that time, we used a model of PAR4 and were still able to successfully develop a series of compounds with biological activity,” Smith said. “This opens up opportunities for structure-based, computer-aided drug design to numerous targets without experimental structures.”
The PAR4 ligands resulting from this screen constitute a novel series with different pharmacology than earlier families of PAR4 ligands. The authors will use the new series to further probe the structure and function of PAR4 and how the ligands themselves are interacting with and inhibiting the receptor. Ultimately, they hope that their work will show other researchers that PAR4, and other receptors like it, are not as hard to target as they might have first thought.
“The ability to block the activity of PAR4 may provide a therapeutic approach to a number of chronic diseases, including heart disease and stroke, as well as inflammatory states such as Alzheimer’s disease,” Hamm said. “This breakthrough is tremendously exciting!”
Go deeper
The paper “Discovery of Protease-Activated Receptor 4 (PAR4)-Tethered Ligand Antagonists Using Ultralarge Virtual Screening” was published in March 2024 in the journal ACS Pharmacology and Translational Science.
Funding
This paper received funding from the National Institutes of Health, the Deutsche Forschungsgemeinschaft, the Alexander von Humboldt Foundation, and the William K. Warren Foundation.