Multisite phosphoregulation of Cdc25 activity refines the mitotic entrance and exit switches Posted August 13th, 2012 by admin
Lucy X. Lua, Maria Rosa Domingo-Sananesb, Malwina Huzarskaa, Bela Novakb, and Kathleen L. Gould a,c,1
+ Author Affiliations
a Department of Cell and Developmental Biology, and
c The Howard Hughes Medical Institute, Vanderbilt University School of Medicine, Nashville, TN 37212; and
b Oxford Centre for Integrative Systems Biology, Department of Biochemistry, University of Oxford, Oxford OX1 3QU, United Kingdom
Edited by Angelika Amon, Massachusetts Institute of Technology, Cambridge, MA, and approved May 4, 2012 (received for review January 26, 2012)
Abstract
Cyclin-dependent kinase 1 (Cdk1) kinase dephosphorylation and activation by Cdc25 phosphatase are essential for mitotic entry. Activated Cdk1 phosphorylates Cdc25 and other substrates, further activating Cdc25 to form a positive feedback loop that drives the abrupt G2/mitosis switch. Conversely, mitotic exit requires Cdk1 inactivation and reversal of Cdk1 substrate phosphorylation. This dephosphorylation is mediated, in part, by Clp1/Cdc14, a Cdk1-antagonizing phosphatase, which reverses Cdk1 phosphorylation of itself, Cdc25, and other Cdk1 substrates. Thus, Cdc25 phosphoregulation is essential for proper G2–M transition, and its contributions to cell cycle control have been modeled based on studies using Xenopus and human cell extracts. Because cell extract systems only approximate in vivo conditions where proteins interact within dynamic cellular environments, here, we use Schizosaccharomyces pombe to characterize, both experimentally and mathematically, the in vivo contributions of Cdk1-mediated phosphorylation of Cdc25 to the mitotic transition. Through comprehensive mapping of Cdk1 phosphosites on Cdc25 and characterization of phosphomutants, we show that Cdc25 hyperphosphorylation by Cdk1 governs Cdc25 catalytic activation, the precision of mitotic entry, and unvarying cell length but not Cdc25 localization or abundance. We propose a mathematical model that explains Cdc25 regulation by Cdk1 through a distributive and disordered phosphorylation mechanism that ultrasensitively activates Cdc25. We also show that Clp1/Cdc14 dephosphorylation of Cdk1 sites on Cdc25 controls the proper timing of cell division, a mechanism that is likely due to the double negative feedback loop between Clp1/Cdc14 and Cdc25 that controls the abruptness of the mitotic exit switch.
Cyclin-dependent kinases (CDKs) are key regulators of the eukaryotic cell cycle. At mitotic entry, the Cdc25 family phosphatases activate Cdk1-CyclinB complexes by removing inhibitory phosphorylations on Cdk1 catalyzed by Wee1 family kinases. Activated Cdk1-CyclinB phosphorylates its substrates and drives mitotic entry (1, 2). Cell cycle modeling showed that a bistable trigger facilitates the switch-like transition between interphase and mitosis (3⇓–5). Bistability ensures that there can only be two stable steady states for the system (interphase or mitosis); it predicts a Cdk1 activity threshold for mitotic entry and a lower activity threshold for mitotic exit, thus giving rise to hysteresis in the system.
Xenopus laevis and human cell extract studies found that the bistable mitotic switch is modulated by at least two feedback loops: the Cdk1-Wee1 double negative feedback loop, in which Cdk1 and Wee1 inactivate one another by phosphorylation, and the Cdk1-Cdc25 positive feedback loop, where Cdk1 phosphorylates and activates Cdc25, while Cdc25 dephosphorylates and further activates Cdk1 (3⇓⇓⇓–7). In addition to a positive or double negative feedback loop, bistability requires an ultrasensitive response of at least one component of a feedback loop (8, 9). It has been proposed that Cdc25 activation and Wee1 inactivation by Cdk1 are ultrasensitive in nature; their activity follows sigmoid signal response curves (rising and decreasing, respectively) as a function of Cdk1 activity (10, 11). In Xenopus egg extracts, ultrasensitivity in Wee1 inactivation is attributed to competition between essential and nonessential Cdk1 phosphosites on Wee1 and between Wee1 and other Cdk1 substrates (6). In addition, Cdk1 multisite phosphorylation of XCdc25C contributes to its ultrasensitive activation (7). Although ex vivo and mathematical models suggest that both Cdk1-Wee1 and Cdk1-Cdc25 feedback loops contribute to the robustness of the mitotic entry switch, perturbation of the feedback loops in vivo in cycling cells has yet to be analyzed.
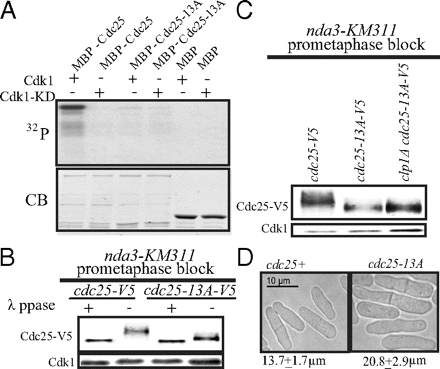
Mitotic exit and the spindle assembly checkpoint may also be modulated by a bistable switch (12⇓–14). In Saccharomyces cerevisiae, Cdc14, a phosphatase that dephosphorylates Cdk1 substrates (15, 16), adds abruptness to the metaphase–anaphase switch by interacting with Securin, a protein that protects sister chromatid separation until anaphase onset in an ultrasensitive positive feedback loop. Cdc14 dephosphorylates Securin to target it for ubiquitylation and degradation. Degradation of Securin activates Separase, which also activates Cdc14 (13). Our laboratory and other groups found that Clp1, the Schizosaccharomyces pombe Cdc14 ortholog, dephosphorylates Cdc25 on Cdk1 phosphosites, and this dephosphorylation correlates with Cdc25 inactivation and degradation (17, 18). Because Cdc25 activates Cdk1, the activity of which inhibits Clp1 activity (19), the interaction between Clp1 and Cdc25 may form a feedback loop that contributes to the mitotic exit switch in S. pombe.
Here, we use S. pombe to further understand how Cdc25 phosphorylation by Cdk1 contributes to the mitotic entry and exit switches in cycling cells. Using this in vivo model, we suggest a mechanism of direct Cdk1 activation and Clp1 inactivation of Cdc25. Also, we find that the Cdk1-Cdc25 positive feedback loop is important for the precision of mitotic entry and maintenance of uniform cell length. Finally, we suggest that the interactions of Clp1, Cdk1, and Cdc25 create a double negative feedback loop that significantly contributes to the robustness of mitotic exit, specifically controlling the timing of cell division.